Abstract
In our research last quarter, we concentrated on identifying antimicrobial peptide (AMP) sequences within microbial communities. Our current study broadens this scope, profiling AMPs across a diverse range of organisms. We aim to refine large datasets to contribute to microbiology improvements through advanced sequence processing techniques. We utilize a Convolutional Neural Network (CNN) and leverage the extensive sequencing data in NCBI’s Reference Sequence Database (O’Leary et al. (2015)) to create detailed AMP profiles for thousands of unique species and uncover new AMPs. The impact of this research is significant within the field of microbiology, offering valuable insights into how cells adapt to manage their local microbial ecosystems. The profiling of AMPs also opens up new directions for alternative solutions/therapeutics, addressing the urgent need for antibiotics that pathogens have not developed resistance against. This research adds to the foundational scientific knowledge of microbial genomics, which is impactful towards several fields like innovation in drug discovery and development.
Intro
Our Quarter 1 project laid the foundation for identifying antimicrobial peptides within microbial communities. As we transition into Quarter 2, our focus shifts towards a broader application of this concept. With the decline of current antibiotic effectiveness due to increasing resistance by harmful bacteria and other pathogens, the search for alternative treatments is more crucial than ever. (World Health Organization (2023)) One promising area of research is the study of antimicrobial peptides (AMPs). AMPs are chains of 2+ amino acids which have some form of antimicrobial activity. The mechanism of action behind each AMP can be extraordinarily varied, making the development of resistance significantly less likely (Yeaman and Yount (2003)). Our project is focused on this exciting potential, looking to map out and understand the AMPs found across different forms of life to better understand how these sequences interact directly with the cells around them.
We were unable to find any prior research that provided insights into how these peptides function as a part of natural immune defenses. We need broader research to really understand how to use them across all forms of life for medical treatments. Our work expands on previous research by applying advanced machine learning techniques to identify AMPs in a wide variety of organisms and to understand their structure and activity on a larger scale.
Our study is built on a large collection of protein data from the RefSeq database (O’Leary et al. (2015)). We apply machine learning methods such as convolutional neural networks (CNNs) and long short-term memory (LSTM) networks to analyze this proteomic information. By focusing on sequenced protein data derived from genetic information, these techniques enable us to find distribution patterns of AMPs across various organisms. While our research will not discover new mechanisms of AMP function, it provides a detailed map of AMP prevalence, offering valuable data for the development of antimicrobial drugs and enhancing our public health strategies against drug-resistant diseases.
Methods
Datasets
The data utilized in this study was sourced from the Reference Sequence (RefSeq) database (O’ Leary et al. (2015)), a comprehensive set of sequences provided by our mentor, Rob Knight. The RefSeq dataset comprised 358,973 files, with each file containing between 4,000 - 6,000 protein sequences derived from genomic data. Data preprocessing involved removing duplicate entries based on the species ID to eliminate redundancy, converting filepaths to a more efficient format for faster processing, and excluding entries with nonexistent protein filepaths to ensure data integrity. This rigorous preprocessing pipeline yielded 62,607 unique species/samples for downstream analysis. To enrich the dataset, full taxonomic lineages for each species were obtained by employing the ncbitax2lin tool (available at https://github.com/zyxue/ncbitax2lin), which leverages the NCBI taxonomy dump Sayers et al. (2019); Schoch et al. (2020), a curated set of names and nomenclature for all organisms represented in the public sequence databases.
The merged lineage dataset contains four overarching superkingdoms (Bacteria, Eukaryotes, Archaea, and Viruses). These superkingdoms are further stratified into phylum, class, order, family, and genus taxonomic ranks. Table 1 presents a subset of the processed species, illustrating the organism name, taxonomic identifier, and taxonomic lineage, while omitting the filepath column for brevity.
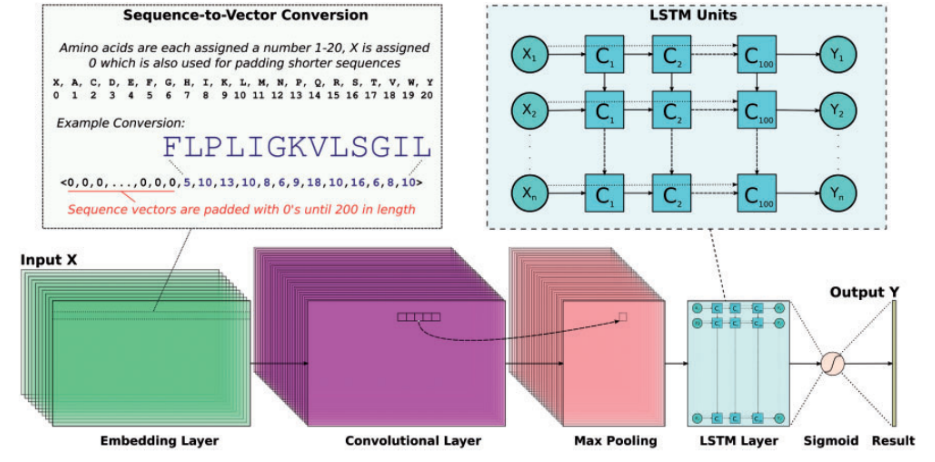
The architecture of our DNN begins by transforming the peptide sequences into numerical vectors of length 200 to fit the longest AMP of the training set at 183 amino acids and the longest non-AMP at 175 amino acids. This is done by first breaking the sequence apart such that each character of the sequence is a single entry in an array. Next we assign an integer to each of the 20 basic amino acid characters and front-pad the sequence with 0’s to make the string 200 characters in length.
Next, these sequences are passed into an embedding layer, which takes as input the length 200 array and organizes it into a smaller vector of length 128. The convolutional layer used in this project was a 1D conv layer (filters = 64, kernel_size = 16, activation="relu"). A convolutional layer acts as a way to store large amounts of information into a small vector while still containing all the necessary information provided to it. In short, a convolutional layer takes a small part of the input and stores small sections as certain values for the output. Do this for the entire dataset and you’re left with a compressed data structure that is easier to handle.
These new sequences of length 16 are passed onto a max pooling layer (pool_length = 5) that uses a sliding window method across the sequence and takes the largest value from it, giving back a representation of the output from the convolutional layer and helps to avoid overfitting. Our model incorporates the use of an LSTM layer (units = 100, unroll = True, stateful = False, dropout = 0.1), which stands for Long Short Term Memory layer. The main purpose of this layer is that it learns long term dependencies of sequential data, making it a perfect fit for peptide sequences. An LSTM layer is a recurrent neural network, meaning that, unlike a traditional neural network, it stores and uses the data it has been passed in as a reference point for the rest of the input. Being able to remember the sequence and its attributes as it gets passed in helps to identify these AMPs. Lastly, the model is tuned with an Adam optimizer with default values and is scored with accuracy using binary cross entropy as our loss function and trained for 10 epochs.
Results
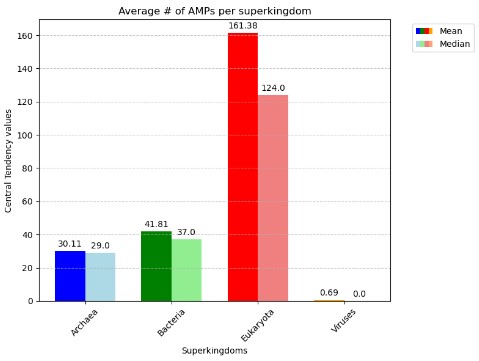
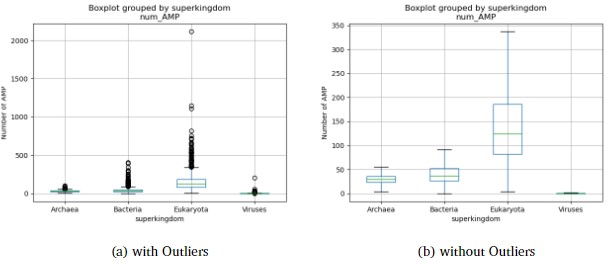
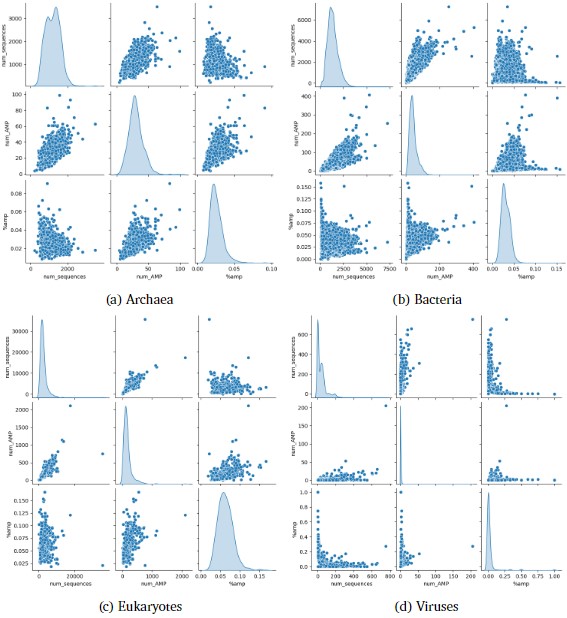
The convolutional neural network model processed a total of 62,604 files, corresponding to an equal number of species, encompassing 66,725,844 amino acid sequences. The percentage of sequences identified as antimicrobial peptides (AMPs) varied across the different superkingdoms, with 2.58% in Archaea, 3.08% in Bacteria, 6.26% in Eukaryota, and 2.83% in Viruses. The number of AMPs per superkingdom divided by the number of samples was 30.1 for Archaea, 41.8 for Bacteria, 161.6 for Eukaryota, and 0.69 for Viruses. Notably, for the Viruses superkingdom, the median percentage of AMPs was 0%, with the 75th percentile at 1.7%, indicating a skewed distribution. The largest outliers from the Eukaryota and Viruses superkingdoms were Triticum dicoccoides with 2,114 AMPs and Pandoravirus inopinatum with 205 AMPs, respectively, as observed in Figure 3. A subset of the identified AMPs was characterized using the AlphaFold protein structure database and UniProt classifications, revealing that a majority were likely membrane proteins. Additionally, 4,251 species, all of which were classified as Viruses, were excluded from the analysis due to the absence of sequences within the specified length range of 10 to 200 amino acids. This exclusion was not based on a general assumption but rather on a comprehensive scan of the results, confirming that 100% of the skipped samples belonged to the Viruses superkingdom.
Discussion
Analysis of Results
Of the four superkingdoms present in the dataset (Figure 2), the eukaryotes exhibited a significantly higher average number of antimicrobial peptides (AMPs) compared to the other superkingdoms, a striking observation that warrants further investigation. This disparity can be attributed to the intricate cellular organization of eukaryotic organisms, which allows for the accommodation of a larger repertoire of AMPs and amino acid sequences within each species. Moreover, as eukaryotes face constant threats from microbial invaders, the elevated presence of AMPs serves as a robust defense mechanism, a plausible evolutionary adaptation to bolster their survival.In Figure 3, we observed that our model consistently identified antimicrobial peptides (AMPs) across large datasets efficiently. This is an improvement over some prior studies. Despite diverse data volumes, our processing times remained stable, showing our model’s potential for high bandwidth applications. Limitations include dependency on accurate AMP labeling and the scope of the training data. Future work could involve refining the model with more datasets and exploring new machine learning techniques to enhance AMP identification and contribute to combating antibiotic resistance.
In contrast, viral species displayed an exceptionally low number of AMPs, with the median value being zero among the 6500+ viral species analyzed. This finding is not surprising when we consider the nature of viruses and their reliance on maintaining the host organism’s viability for their propagation. The presence of numerous AMPs could potentially disrupt the host’s homeostasis, thereby reducing the chances of viral proliferation and jeopardizing their survival strategy.
The analysis also extends to the specific properties of the AMPs themselves, unveiling intriguing insights. Many of these peptides can be characterized as membrane proteins, a finding that aligns with their functional role of mediating cell-environment interactions, positioning them as the first line of defense against harmful microbes. Consequently, their antimicrobial properties serve as a strategic adaptation for cellular protection.
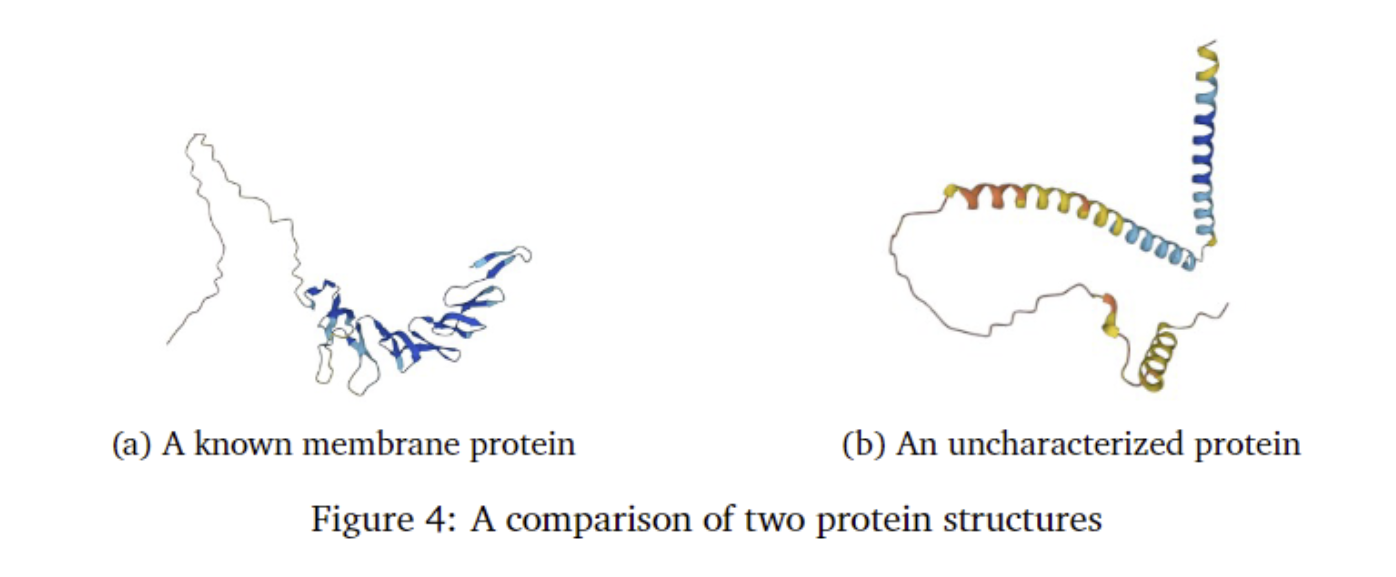
Furthermore, the characterized membrane proteins identified as AMPs can shed light on uncharacterized proteins that exhibit antimicrobial properties. A prime example is illustrated in the figure below. While the protein on the right is uncharacterized by biologists, it shares striking similarities with the known membrane protein on the left. Both are classified as AMPs, and both exhibit a distinct structural pattern, featuring a densely populated amino acid head region that tapers into a long tail-like structure. Leveraging this knowledge, we can reasonably hypothesize that the uncharacterized protein on the right is also a membrane protein used by the cell for defensive purposes, given their structural resemblance.
Conclusion
The knowledge and results obtained from this experiment have paved the way for numerous potential future research avenues. One promising direction lies in investigating the differences in AMP clusters and their distribution across various taxonomic groups. Such an endeavor could prove invaluable in understanding the transitive properties of certain AMPs and their potential applications in combating diverse microbial threats.Notably, the eukaryotic organisms have a significantly higher rate of AMPs compared to other kingdoms. These peptides give insight into the ways eukaryotes manage the microbial communities around them. Delving deeper into the properties of specific AMPs can shed further light on our current understanding of these molecules and possibly lead to the creation of new classes of antibiotics.
The results from this study also highlight the potential for identifying and classifying the uncharacterized AMPs that our model has uncovered. While many AMPs could be membrane proteins, a significant number originate from species that have not been extensively studied, hindering their proper characterization. Building upon the findings illustrated in Figure 4, it may be prudent to develop a novel machine learning model to analyze the amino acid sequences that compose these uncharacterized AMPs and attempt to find relations to characterized AMPs.
Limitations
Due to time and computational constraints, our analysis was limited to a single example per species from the large datasets we had access to. No duplicate entries were included in the process. While we believe that this limitation does not significantly impact the overall output, it is worth noting that there may be specific edge cases or instances where a sample from a different part of the organism could have been more representative.Additionally, our findings relied on the accuracy of the employed model. Currently, we utilize a 99% confidence metric to determine whether a sequence is classified as an AMP. Although the results thus far do not indicate a need for adjustment, it is important to acknowledge that even a slight modification, such as using a 95% confidence interval, could lead to substantial changes in the outcome.
Future Research
Future research aims to establish taxonomic relationships to identify the most prevalent AMPs across species. This work will enhance our understanding of common AMPs within taxonomic clusters and clarify how organisms adapt to their environments. Identifying AMP clusters in various species, we seek insights into their function and evolutionary importance. This study will reveal how species and taxonomies control their microbial populations, highlighting AMPs’ key role in microbial adaptationReferences
O’Leary, Nuala A, Mathew W Wright, J Rodney Brister, Stacy Ciufo, Diana Haddad, Rich McVeigh, Bhanu Rajput, Barbara Robbertse, Brian Smith-White, Danso AkoAdjei, Alexander Astashyn, Azat Badretdin, Yiming Bao, Olga Blinkova, Vyacheslav Brover, Vyacheslav Chetvernin, Jinna Choi, Eric Cox, Olga Ermolaeva, Catherine M Farrell, Tamara Goldfarb, Tripti Gupta, Daniel Haft, Eneida Hatcher, Wratko Hlavina, Vinita S Joardar, Vamsi K Kodali, Wenjun Li, Donna Maglott, Patrick Masterson, Kelly M McGarvey, Michael R Murphy, Kathleen O’Neill, Shashikant Pujar, Sanjida H Rangwala, Daniel Rausch, Lillian D Riddick, Conrad Schoch, Andrei Shkeda, Susan S Storz, Hanzhen Sun, Francoise Thibaud-Nissen, Igor Tolstoy, Raymond E Tully, Anjana R Vatsan, Craig Wallin, David Webb, Wendy Wu, Melissa J Landrum, Avi Kimchi, Tatiana Tatusova, Michael DiCuccio, Paul Kitts, Terence D Murphy, and Kim D Pruitt. 2015. “Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation.” Nucleic Acids Res 44(D1): D733–45Sayers, Eric W, Mark Cavanaugh, Karen Clark, James Ostell, Kim D Pruitt, and Ilene Karsch-Mizrachi. 2019. “GenBank.” Nucleic Acids Res. 47(D1): D94–D99
Schoch, Conrad L, Stacy Ciufo, Mikhail Domrachev, Carol L Hotton, Sivakumar Kannan, Rogneda Khovanskaya, Detlef Leipe, Richard Mcveigh, Kathleen O’Neill, Barbara Robbertse, Shobha Sharma, Vladimir Soussov, John P Sullivan, Lu Sun, Seán Turner, and Ilene Karsch-Mizrachi. 2020. “NCBI Taxonomy: a comprehensive update on curation, resources and tools.” Database (Oxford) 2020
Veltri, Daniel, Uday Kamath, and Amarda Shehu. 2018. “Deep learning improves antimicrobial peptide recognition.” Bioinformatics 34(16): 2740–2747. [Link]
World Health Organization. 2023. “Antimicrobial resistance.” Nov. [Link]
Yeaman, Michael R, and Nannette Y Yount. 2003. “Mechanisms of antimicrobial peptide action and resistance.” Pharmacol Rev 55(1): 27–55